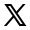
Con motivo del Día Mundial de las Enfermedades Raras, desde la Sociedad Española de Investigación Ósea y del Metabolismo Mineral (SEIOMM) hemos realizado una serie de entrevistas con especialistas para dar visibilidad a distintas enfermedades raras que afectan al hueso y al metabolismo mineral.
A continuación recogemos estas entrevistas para conocer mejor en qué consisten estas patologías, cuáles son sus síntomas, cómo se diagnostican y cuál es la importancia de su tratamiento.
Entrevista a Àngels Martínez Ferrer

Reumatóloga · Hospital Universitari Dr. Peset (València)
Tesorera de la Junta Directiva de la SEIOMM
¿En qué consiste la osteogénesis imperfecta?
La osteogénesis imperfecta es un trastorno hereditario que provoca fragilidad ósea debido a defectos en el colágeno tipo I, causados principalmente por mutaciones en los genes COL1A1 o COL1A2.
¿Qué síntomas puede presentar el paciente?
Su característica clínica más típica es la fragilidad ósea, que provoca fracturas frecuentes ante traumatismos mínimos o espontáneos —especialmente durante la infancia—, dándole el nombre de enfermedad de los huesos de cristal.
La gravedad y otros síntomas, como escleras azules, dentinogénesis imperfecta o hipoacusia, varían según el tipo y severidad del trastorno.
¿Cómo se diagnostica?
El diagnóstico de la osteogénesis imperfecta es fundamentalmente clínico, basándose en la recurrencia de fracturas y la presencia de rasgos característicos como las escleras azules.
Para confirmarlo, se emplean radiografías y densitometrías que evalúan la fragilidad ósea, aunque el método definitivo es el estudio genético para identificar mutaciones en el colágeno.
Importancia del tratamiento
El abordaje terapéutico es fundamental para optimizar la densidad mineral ósea y reducir la morbilidad asociada a la fragilidad esquelética.
Su importancia radica en la prevención de fracturas recurrentes y la corrección de deformidades mediante un manejo multidisciplinar que combina fisioterapia, osteosíntesis intramedular y una intervención farmacológica especializada.
Esta se basa en el uso de bisfosfonatos para inhibir la resorción ósea, aunque actualmente se incorporan otras opciones como el denosumab o la teriparatida, adaptando la elección según el perfil y la edad del paciente.
Entrevista a Carolina Tornero Marín

Servicio de Reumatología · Hospital Universitario La Paz
¿En qué consiste la hipofosfatemia ligada al cromosoma X (XLH)?
La hipofosfatemia ligada al cromosoma X (XLH; OMIM 307800) es un trastorno genético poco frecuente causado por variantes con pérdida de función en el gen regulador del fosfato con homología a endopeptidasas (PHEX).
Esto da lugar a un aumento de las concentraciones circulantes de factor de crecimiento fibroblástico 23 (FGF23), que altera la reabsorción renal de fosfato y provoca su pérdida, dando lugar a hipofosfatemia crónica y osteomalacia.
¿Qué síntomas puede presentar el paciente?
La hipofosfatemia ligada al cromosoma X (XLH) se caracteriza por la presencia de talla baja desproporcionada, deformidades en extremidades de carga, osteomalacia con fracturas o pseudofracturas, dolor óseo y articular crónico, daño articular precoz, entesopatía y debilidad muscular.
Son frecuentes las alteraciones dentales y pueden presentarse complicaciones renales, como nefrocalcinosis y urolitiasis, neurológicas y pérdida auditiva.
La enfermedad se asocia a limitación funcional progresiva, fatiga y deterioro de la movilidad y de la función física, condicionando un impacto significativo en la calidad de vida de los pacientes.
¿Cómo se diagnostica?
El diagnóstico se basa en la integración de la evaluación clínica, los hallazgos de laboratorio, que incluyen la presencia de hipofosfatemia crónica, pérdida renal de fosfato en ausencia de otras causas y niveles de FGF23 elevados o inapropiadamente normales, y los radiológicos.
La XLH se confirma mediante la identificación de una variante patogénica o probablemente patogénica en el gen PHEX y resulta de especial utilidad cuando no es evidente un patrón de herencia dominante ligado al X en la familia.
Importancia del tratamiento
El tratamiento de la XLH en la edad adulta es fundamental para reducir los síntomas, disminuir la carga de enfermedad y limitar la progresión de complicaciones estructurales y sistémicas a largo plazo.
Su abordaje debe realizarse en el contexto de un manejo multidisciplinar.
El tratamiento convencional en adultos sintomáticos incluye la administración de sales de fosfato y vitamina D activa.
Más recientemente, se ha incorporado al arsenal terapéutico burosumab, un anticuerpo monoclonal dirigido contra FGF23, que abre un nuevo paradigma en el enfoque terapéutico de la enfermedad.
Entrevista a Arantxa Conesa Mateos

Jefa de Sección de Reumatología · Universidad Jaume I (Castellón)
Profesora asociada UJI
¿En qué consiste la enfermedad de Paget?
Se caracteriza por ser un trastorno focal del remodelado óseo secundario a una hiperactividad osteoclástica seguida de una formación ósea desorganizada y anárquica, que conlleva a un hueso estructuralmente menos resistente, con deformidades y mayor riesgo de complicaciones mecánicas y neurológicas.
¿Qué síntomas puede presentar el paciente?
Las manifestaciones más frecuentes son el dolor, deformidad ósea y fracturas por fragilidad, a pesar de que la mayor parte de los pacientes son asintomáticos (40-70%) y son diagnosticados incidentalmente al realizar pruebas diagnósticas de laboratorio o imagen por otro motivo.
¿Cómo se diagnostica?
El diagnóstico se basa en hallazgos radiológicos característicos y en marcadores bioquímicos del remodelado óseo, como la fosfatasa alcalina total (FAT).
Importancia del tratamiento
El tratamiento de la enfermedad se basa en fármacos antiresortivos.
Su importancia radica en que permite suprimir la actividad osteoclástica aumentada, aliviar el dolor óseo, normalizar los marcadores bioquímicos (como la fosfatasa alcalina), prevenir deformidades y fracturas, y reducir el riesgo de complicaciones neurológicas, articulares y, en casos raros, transformación sarcomatosa.
Entrevista a Diana Ovejero

Especialista en endocrinología · Hospital del Mar Research Institute, CIBERFES
Unidad de Investigación Músculo-Esquelética
¿En qué consiste la displasia fibrosa?
La displasia fibrosa es una enfermedad mosaica secundaria a mutaciones activantes del gen GNAS que da lugar al desarrollo de lesiones óseas en las que el hueso normal es sustituido por un tejido fibroso anómalo.
El síndrome de McCune-Albright es la asociación de estas lesiones óseas con manchas cutáneas y endocrinopatías como la pubertad precoz, el hipertiroidismo o la acromegalia, entre otras.
¿Qué síntomas puede presentar el paciente?
Los pacientes con lesiones óseas únicas pueden ser completamente asintomáticos, pero dependiendo del número de lesiones y de su localización pueden dar lugar a deformidades, dolor óseo, trastornos en la marcha y fragilidad esquelética.
También pueden producir compresión nerviosa, incluyendo sordera y afectación visual en casos de afectación craneofacial.
En los pacientes con síndrome de McCune-Albright se añaden los síntomas derivados de las endocrinopatías que padezcan.
¿Cómo se diagnostica?
En casos de lesiones de displasia fibrosa aislada, lo ideal es realizar una biopsia y secuenciar el gen GNAS.
En aquellos pacientes con más de una lesión, se considera que se puede establecer el diagnóstico si las lesiones presentan un aspecto radiológico característico y son hipercaptantes en la gammagrafía ósea.
Importancia del tratamiento
El abordaje está completamente supeditado a las manifestaciones de cada paciente, que son variables dada la naturaleza mosaica de la enfermedad.
Puede requerir cirugía ortopédica y maxilofacial, oftalmológica, rehabilitación, suplementación de fosfato y vitamina D, y tratamiento endocrinológico específico.
Entrevista a Manuel Muñoz Torres

Catedrático de Medicina · Universidad de Granada
Hospital Universitario San Cecilio (Granada)
¿En qué consiste la hipofosfatasia?
La hipofosfatasia es una enfermedad genética rara caracterizada por una deficiencia de la enzima fosfatasa alcalina, necesaria para la mineralización adecuada de los huesos y otras funciones metabólicas.
¿Qué síntomas puede presentar el paciente?
Existen formas pediátricas, las más graves, que se manifiestan con signos de raquitismo grave y síntomas neurológicos.
Las formas del adulto pueden originar fracturas por alteración de la mineralización, dolores articulares y debilidad muscular. Algunas son asintomáticas.
¿Cómo se diagnostica?
Se diagnostica comprobando que los valores en sangre de fosfatasa alcalina total están bajos de forma mantenida y aumentan lo que denominamos sustratos (fundamentalmente piridoxal fosfato o PLP).
El estudio genético permite identificar la mutación del gen responsable.
Importancia del tratamiento
Las formas más graves se pueden tratar con terapia enzimática de sustitución.
Las formas leves se tratan con medidas de soporte como ortesis, antiinflamatorios o cirugía ortopédica cuando es necesario.
Es importante no administrar bisfosfonatos porque la mineralización ósea puede empeorar.





